Etiquetas
coronavirus, David Baltimore, Premio Nobel, Retrotranscriptasa, retrovirus, Virus ARN

Los coronavirus de Wuhan son genéticamente “retrovirus”, tienen como material genético ARN.
David Baltimore es un biólogo estadounidense Premio Nobel de Fisiología o Medicina en 1975, su trabajo consistió en el hallazgo de que el ARN puede ser transcrito por una retrotranscriptasa a ADN, una enzima que se utiliza para replicarse en las células humanas. Importante en el desarrollo de antivirales, clave en las infecciones virales.
Ha revolucionado el campo de la biología molecular, con aplicaciones diversas: detección de enfermedades, terapias de fármacos y en la identificación de perfiles de genes celulares cuya expresión es modulada en respuesta a una infección producida por patógenos.
Baltimore también ha hecho una clasificación de los virus agrupados en grupos dependiendo su tipo de genoma ADN o ARN, monocatenario o bicatenario y su método de replicación, clasificar los virus según su genoma implica que los que quedan encuadrados en la misma categoría se comportarán básicamente de la misma manera, facilitando las investigaciones.

Existen dos clasificaciones de los virus que están autorizadas, por el Comité Internacional de Taxonomía de Virus (estos dos métodos de clasificación no son antagónicas, pueden integrarse entre sí, pues la clasificación del ICTV incluye algunos criterios de la clasificación de Baltimore):
a) La Clasificación de Baltimore, basada en el tipo de ácido nucleico de los virus (ADN o ARN) y su modo de expresión génica.
b) La clasificación del Comité Internacional de Taxonomía de Virus (ICTV), similar al sistema de clasificación de los seres vivos: orden, familia, subfamilia, género y especie:
GRUPO I: Virus ADN bicatenario (Virus ADNbc o Virus dsDNA)
GRUPO II: Virus ADN monocatenario (Virus ADNmc o Virus SSDNA)
GRUPO III: Virus ARN bicatenario (Virus ARNbc o Virus dsRNA)
GRUPO IV: Virus ARN monocatenario positivo (Virus ARNmc+ o Virus (+) ssRNA)
GRUPO V: Virus ARN monocatenario negativo (Virus ARNmc- o Virus (-) ssRNA)
GRUPO VI: Virus ARN monocatenario retrotranscrito (Virus ARNmcRT o Virus ssRNA-RT)
GRUPO VII: Virus ADN bicatenario retrotranscrito (Virus ADNbcRT o Virus deDNA-RT)
En su concepción original, la clasificación de Baltimore incluía seis clases de genoma vírico, más adelante se han descubierto el genoma de los hepadnavirus (virus de la hepatitis B) para los que se añadió una séptima clase.
El “dogma central de la biología” es que el que código genético se encuentra en el DNA, se transcribe a un RNA mensajero cuya información se traduce a proteínas, los ladrillos de la vida. El estudio de la reproducción de los “retrovirus” poseen la capacidad de transformar células normales sintetizando DNA viral a partir de su genoma RNA mediante una reacción catalizada por la enzima viral “Transcriptasa reversa”, una polimerasa de DNA dependiente de RNA
Revolucionando el campo de la biología molecular, la replicación de los retrovirus es incompatible con el dogma central.
La clasificación de los virus es el proceso de nombrar los virus y colocarlos en un sistema taxonómico, la clasificación de Baltimore de los virus, se basa en el método de síntesis viral del ARN mensajero (ARNm), no supone que es filogenética, ya que los virus no comparten un origen común.
El interés por la virología se inició para controlar las infecciones virales que afectaban a la agricultura, ganadería y la salud humana desde épocas antiguas, el interés por entender en los tiempos actuales los procesos celulares y moleculares.
Los coronavirus es un ejemplo de lo que se denomina zoonosis: enfermedades de los animales que pueden pasar al ser humano, los análisis los relacionan con el grupo 2B de los coronavirus, dentro de la misma familia que el SARS. Más del 60% de las nuevas infecciones emergentes o reemergentes, como son estos casos de los coronavirus son de origen animal, su extensión quizá se deba a nuestra capacidad de movernos por el planeta que a las características de los propios virus. Accidentalmente “han saltado” al ser humano causando un síndrome respiratorio.
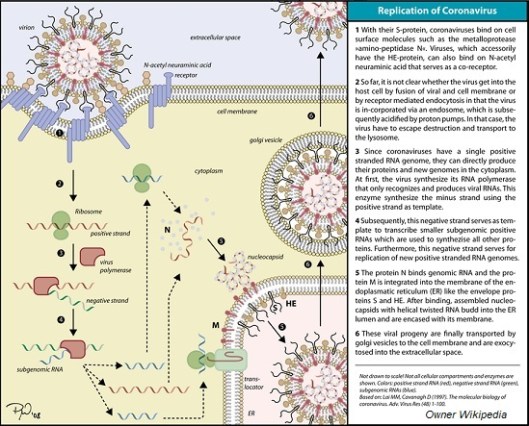
Son un grupo de virus de la clase IV (según la clasificación de Baltimore) con genoma compuesto por una sola hebra de ARN sentido positivo. Rodeados por una envoltura con proteínas que se proyectan hacia el exterior. Se han aislado en gran cantidad de animales: perros, gatos, cerdos, vacas, aves, roedores, murciélagos, camellos. En humanos la infección por coronavirus es frecuente y causan enfermedades leves o moderadas del tracto respiratorio superior, conjuntivitis o trastornos gastrointestinales. Desde 2003 se han descrito otros coronavirus de origen animal que han infectado al ser humano causando síndromes respiratorios, son los virus SARS y MERS.
Bibliografía:
David Baltimore Nobel Prize
https://www.nobelprize.org/prizes/medicine/1975/baltimore/facts/
David Baltimore “Viruses, Polymerases and Cancer”, Nobel Lecture. NobelPrize.org. Nobel Media AB, 2020
https://www.nobelprize.org/prizes/medicine/1975/baltimore/lecture/
David Baltimore iBiology
David Baltimore, «Presentación introducción a los virus Transcriptasa inversa» iBiology
Baltimore D, “Expression of animal virus genomes”, Bacteriol Rev, 1971
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC378387/
Baltimore´s Home Rockefeller University
http://biology.caltech.edu/Members/Baltimore