Las enfermedades raras, tradicionalmente huérfanas, han pasado a tener una gran importancia en la última década. Incluidas las de origen genético, son aquellas enfermedades que afectan a un pequeño número absoluto de personas o a una proporción reducida de la población. Son crónicas y con frecuencia progresivas, degenerativas, presentándose con elevada morbilidad y mortalidad. Conllevan una gran cantidad de síntomas que afectan a distintas capacidades, así los síntomas son en el plano físico, sensorial, mental y de comportamiento, los síntomas pueden afectar a cada persona de manera diferente. Una enfermedad para ser considerada rara no debe afectar a más de 24.000 personas.
Las enfermedades raras son graves y bastante limitantes, uno de cada tres afectados pierde la autonomía personal, una carga para los familiares de los pacientes.
Dos de cada tres enfermedades raras se manifiestan antes de que el niño cumpla dos años. Suelen aparecer durante la primera infancia. Entre el 50 y el 75% afectan a la infancia y un 30% de los pacientes mueren antes de los 5 años. El retraso en el diagnóstico, de 5 a 7 años de promedio, los tratamientos inexistentes o inadecuados, dificultades sociales por el empobrecimiento económico y la discriminación global, hacen que la solución tenga que ser integral.
Afectan a una proporción muy pequeña de la población (1 de cada 2.000 personas en Europa), en la actualidad hay descritas más de 6.000 enfermedades raras, con una repercusión global importante, entre un 6 y 8% de la población,
· en todo el mundo 300 millones de personas
· en Europa entre 27 y 36 millones de personas
· en EEUU unos 30 millones de personas
· en España más de 3 millones
En España hay 50 enfermedades raras que afectan únicamente a algunos miles de personas, unas 500 de ellas sólo en varios centenares de personas y el resto hasta llegar a las 7.000 enfermedades afectarían sólo a decenas de personas.
La enfermedad rara más frecuente en España es el síndrome de aceite tóxico, afecta a casi 15.000 personas, su origen a principios de los 80, con la intoxicación masiva por la ingesta de aceite de colza desnaturalizado.
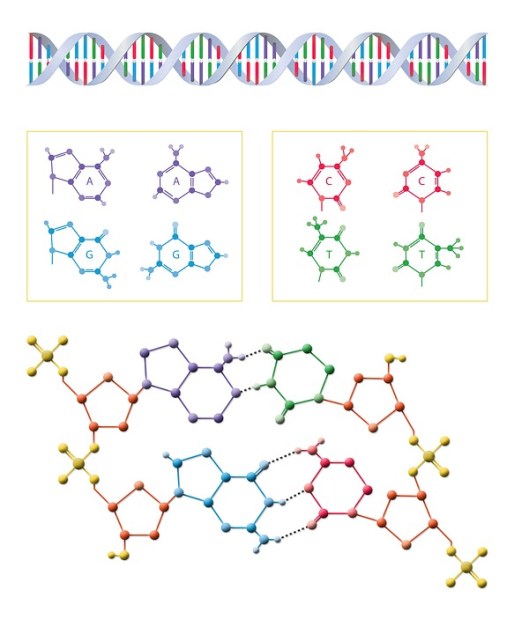
Muchas de las enfermedades raras son de carácter hereditario, conocer qué genes están implicados en una enfermedad rara permite, poder llevar a cabo un diagnóstico temprano y plantear posibles tratamientos para la enfermedad. El desarrollo de técnicas de secuenciación del genoma humano y analizar la parte codificante del mismo (exoma), ha ampliado la posibilidad de identificar mutaciones y sus genes.
El diagnóstico de las enfermedades raras se suele producir con un retraso medio de cinco años. Es importante estudiar la historia familiar para conocer el patrón de herencia y saber las posibilidades de riesgo. La asesoría genética es importante en estas personas con altas posibilidades: “diagnósticos presintomáticos”, que se pueden realizar en las unidades genéticas de los hospitales, derivados de atención primaria.
La definición de medicamento huérfano cambia según los estamentos. El Reglamento (CE) 141/2000 del Parlamento Europeo y del Consejo de la Unión Europea(UE), de 16 de diciembre de 1999, lo define: “una medicina a) para tratar una enfermedad que amenaza la ida del paciente o la debilita de forma crónica, b) que no afecta a más de 5 personas por 10.000 o para la cual se espera un bajo retorno de inversión si no se ofrece un incentivo adicional y c) para el cual se carece de tratamiento alternativo o el nuevo medicamento brinda beneficios adicionales a los pacientes comparado con los tratamientos disponibles “. La autorización de comercialización corre a cargo, del Comíté para Productos Medicinales Huérfanos y del Comité para Productos Medicinales Humanos. Desde 2005, los medicamentos huérfanos sólo pueden obtener la autorización de comercialización mediante un procedimiento centralizado en la EMEA. Se sabe poco de la fisiopatología de estas enfermedades y es difícil reclutar a suficientes pacientes para realizar ensayos clínicos. La EMEA (European Medicines Agency) ha autorizado la comercialización de 47 medicamentos huérfanos y entorno a 1.000 se encuentran en fase de investigación.
En España, algunos centros, como:
- el Instituto de Salud Carlos III y el CIEMAT (Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas) llevan a cabo estudios sobre enfermedades raras
- el CIBERER (Centro de Investigación Biomédica en Red de Enfermedades Raras), coordina a los grupos de investigación que trabajan en estas patologías desde diferentes comunidades autónomas
- ORPHANET es el portal de información de referencia de enfermedades raras y medicamentos huérfanos (inventario, listado de medicamentos, directorio de centros expertos, laboratorios clínicos, proyectos, ensayos clínicos, plataformas tecnológicas y asociaciones de pacientes).
- SEAGen es la Sociedad Española de Asesoramiento Genético, proporciona formación continua al profesional médico.
El informe de la Comisión Nacional de Enfermedades Raras del gobierno de EEUU en 1989 fue el que por primera vez llamó la atención pública. El Consejo de Europa, (2009/C151/02) señaló 7 recomendaciones cuyas líneas de acción se dirigen a la mejora de su reconocimiento y visibilidad, al apoyo de planes nacionales de Estados Miembros y al fortalecimiento de la cooperación y coordinación. En España la Estrategia en Enfermedades Raras del Sistema Nacional de Salud presenta 7 grandes líneas de actuación (indicadores de salud, prevención, detección precoz, atención socio-sanitaria, terapias, investigación e información).
El 29 de febrero se celebra el día Mundial de las Enfermedades Raras.
Bibliografía:
- Salvador Peiró; Carlos Campillo Artero; “Enfermedades raras, medicamentos huérfanos: el valor de la orfandad” http://www.academia.edu/27994274/Enfermedades_raras_medicamentos_huerfanos_el_valor_de_la_orfandad
· OMS, Boletín informativo
Volumen 90, nº 6, junio 2012
· Jules J. Berman; “Enfermedades raras y medicamentos huérfanos: Claves para comprender y tratar las enfermedades comunes”Ed. Elsevier,2015
Links relacionados:
· ministerio de sanidad: enfermedades raras
https://www.msssi.gob.es/ciudadanos/asocEnfermosYFamiliares/enfermedadesRaras.htm
· feder
fEDERACION ESPAÑOLA DE ENFERMEDADES RARAS
· FEDERACIÓN ESPAÑOLA ENFERMEDADES METABÓLICAS HEREDITARIAS
· ORPHANET
PORTAL DE ENFERMEDADES RARAS
http://www.orpha.net/consor4.01/www/cgi-bin/?lng=ES
· oms enfermedades raras
http://www.who.int/bulletin/volumes/90/6/12-020612/es/
· EURODIRS
ORGANIZACIÓNes europeas de Enfermedades raras
https://www.eurordis.org/es/content/federaciones-de-enfermedades-raras
· Lista de enfermedades raras
https://www.hon.ch/HONselect/RareDiseases/index_sp.html
· ema
medicamentos de enfermedades raras
https://www.eupati.eu/es/registro/comites-de-la-ema-comite-de-medicamentos-huerfanos-comp/
· CIBERER
centro de investigación biomédica en red, enfermedades raras
· INSTITUTO GENÉTICA Y GENÓMICA FUNDACIÓN JIMENEZ DÍAZ
https://www.fjd.es/iis_fjd/es/areas-grupos-investigacion/genetica-genomica
· seagen
sociedad española de asesoramiento genético
· IMEGEN TEST GENÉTICOS
· GENYCA LABORATORIO DE ENFERMEDADES RARAS
http://www.genyca.es/analisis-geneticos/enfermedades-raras/